
Unkind cuts - proteolysis and proteomics
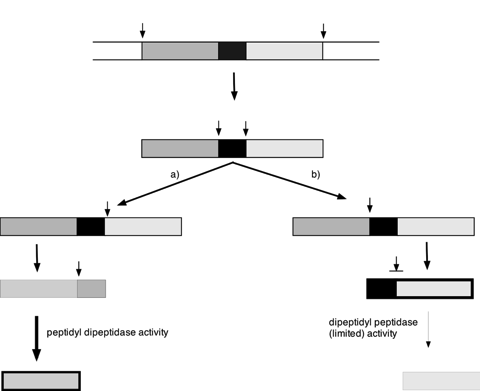
Virtually all mass spectrometric-based methods for quantitative proteomics are at the peptide level, whether label-mediated or label-free. Absolute quantification in particular is based on the measurement of limit peptides, defined as those peptides that cannot be further fragmented by the protease in use. Complete release of analyte and (stable isotope labelled) standard ensures that the most reliable quantification data are recovered, especially when the standard peptides are in a different primary sequence context, such as sometimes occurs in the QconCAT methodology. Moreover, in label-free methods, incomplete digestion would diminish the ion current attributable to limit peptides and lead to artefactually low quantification data. It follows that an essential requirement for peptide-based absolute quantification in proteomics is complete and consistent proteolysis to limit peptides. In this paper we describe strategies to assess completeness of proteolysis and discuss the potential for variance in digestion efficiency to compromise the ensuing quantification data. We examine the potential for kinetically favoured routes of proteolysis, particularly at the last stages of the digestion, to direct products into ‘dead-end’ mis-cleaved products.