Research
In which wild hosts do pathogens hide? Where might novel pathogens come from? What attributes might these pathogens have? These are the interlinked questions I aspire to solve. To date I have made significant inroads to finding the answers by integrating machine-learning, complex-networks, and big data. In the near future, I aim to transform my research from being strictly computational, to becoming verifiable both in the lab and the field.
Multi-perspective prediction of host-pathogen associations
In order to answer questions such as: which hosts might harbour the next pandemic coronavirus? We need to approach the problem from multiple perspectives:
• By only using the pathogens’ data, we would miss key aspects of the hosts and might not be able to pinpoint the exact species affected by the pathogen.
• A solution developed solely from the hosts’ perspective would miss the intricacies of pathogens.
• Utilising only the networks linking hosts and pathogens, we might not be able to identify the drivers determining the hosts of each pathogen.
Hence, I developed innovative methodologies whereby these elements are considered individually, then recombined to increase predictive power
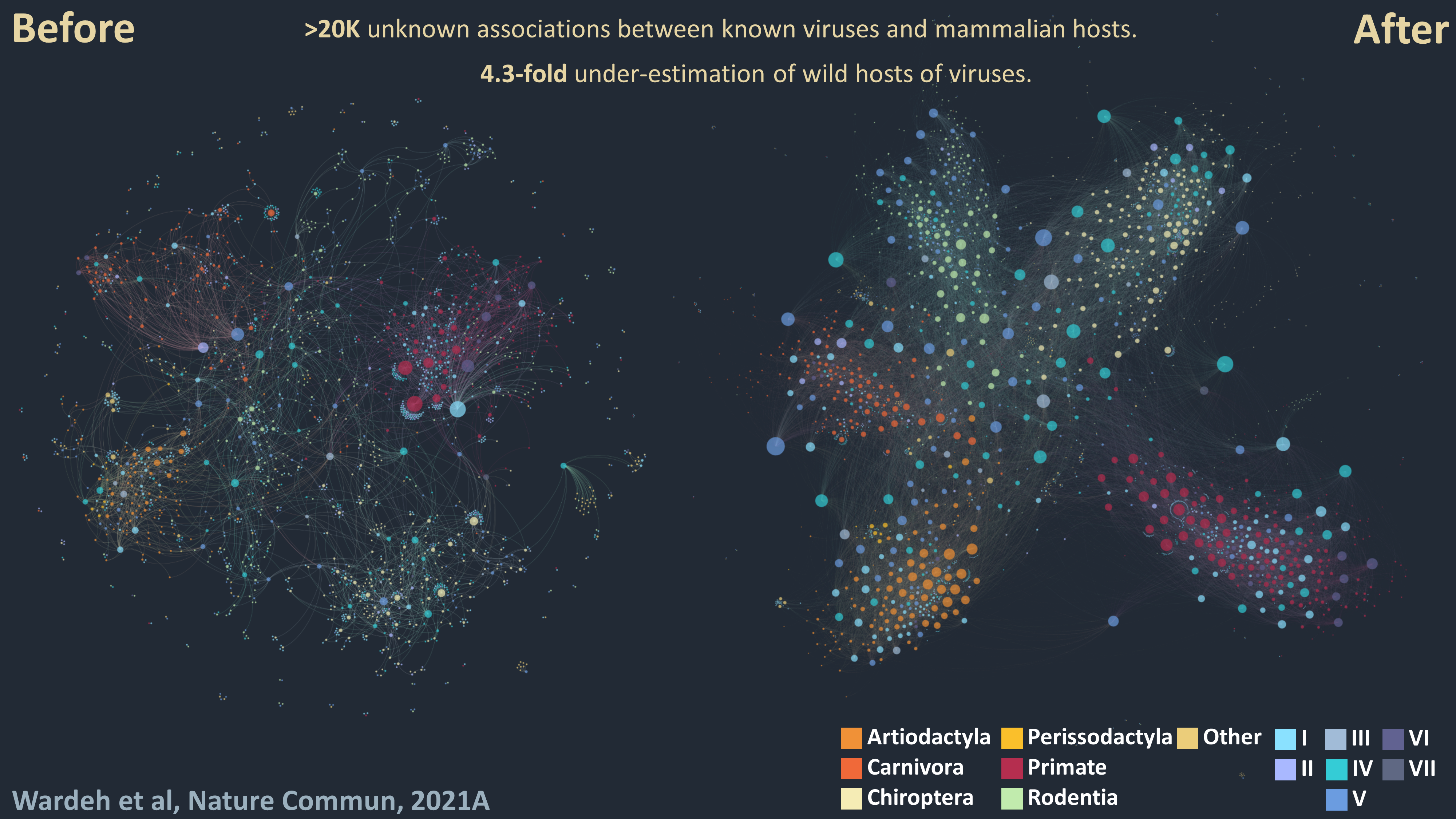
1 - Divide-and-conquer: Our knowledge of viral host ranges remains limited. Completing this picture by identifying unknown hosts of known viruses can help identify and mitigate zoonotic and animal-disease risks. To address this knowledge-gap I developed a divide-and-conquer approach which separates viral, mammalian and network features into three unique perspectives, each predicting associations independently to enhance predictive power. My approach predicts >20,000 unknown associations between known viruses and mammalian hosts, suggesting that current knowledge underestimates the number of associations in wild and semi-domesticated mammals by a factor of 4.3, and the average mammalian host-range of viruses by a factor of 3.2.
2 - Predicting hosts in which coronaviruses recombine: Novel pathogenic coronaviruses – such as SARS-CoV and probably SARS-CoV-2 – arise by homologous recombination between co-infecting viruses in a single cell. Identifying possible sources of novel coronaviruses therefore requires identifying hosts of multiple coronaviruses; however, most coronavirus-host interactions remain unknown. By deploying a meta-ensemble of similarity learners from three complementary perspectives (viral, mammalian and network), I was able to predict which mammalian species are hosts of multiple coronaviruses. The results indicated that there are 11.5-fold more coronavirus-host associations, over 30-fold more potential SARS-CoV-2 recombination hosts, and over 40-fold more host species with four or more different subgenera of coronaviruses than have been observed to date. These results demonstrate the large underappreciation of the potential scale of novel coronavirus generation in animals. They also enable the identification of high-risk species for coronavirus surveillance – a research strand I am currently developing with collaborators across IVES.

Big Data approaches to identifying potential sources of emerging pathogens
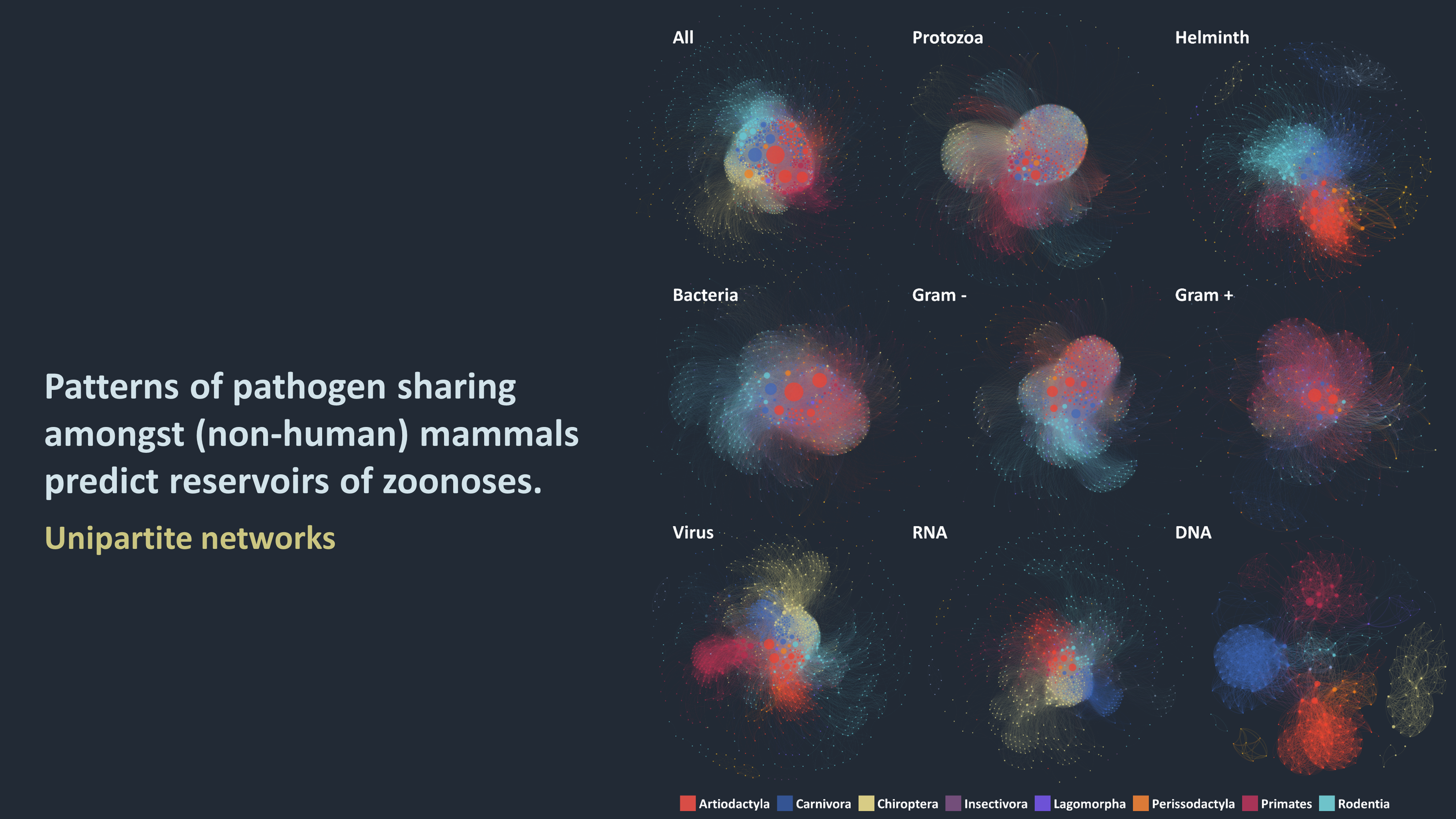
Emerging infectious diseases continue to pose major threats to humans, animals and plants. Recent years have seen significant outbreaks of several emerging diseases, ranging from the well-known (Ebola and Olive quick decline syndrome), to the previously little known (Zika), to the entirely novel (Schmallenberg), to name but a few. It is well established that the ability of a pathogen to infect multiple hosts, particularly hosts in different taxonomic orders or wildlife, is a risk factor for emergence in human and livestock pathogens. Emerging wild-life diseases have also been linked to 'spill-overs' from humans or domesticated animals. Despite the importance of cross-species disease transmission, there has been relatively little attention paid to which species are the most important sources cross communities (e.g., zoonotic, wild-life to domestic, plants to other kingdoms), which are the most prolific vectors, how those species acquired the pathogens, and by what means the diseases entered new species or populations. A major reason for this limited understanding is the lack of comprehensive data on the pathogens in animal and plant populations and, in most cases, poorly documented information on how they are transmitted, including to humans.
At the heart of my research are ecological networks: where nodes represent host or pathogen species (or strain), and links illustrate various interactions between those nodes (such as two hosts sharing a pathogen, or a virus infecting a host). These networks are both tools to depicting indirect and potentially complex interactions (such as transmission cycles of vector-borne pathogens), and an arena of interdisciplinary and active research. I use those networks to advance our understanding of:
1. What are the characteristics of the networks that connect species via shared pathogens? How central are humans and their domesticated animals and crops in these networks and which other species are each of those communities most closely connected to?
2. What is the role of different pathogen transmission routes on the nature of these networks? Are the potential species-to-species transmission pathways different for direct, food-borne, water-borne and vector-borne pathogens?
3. What factors determine the host ranges of pathogens? Are host species more likely to become exposed to pathogens that infect a wide range of species? From species that are closer to them genetically? Or from those species with which they often interact?
4. What are we missing? Given the networks, transmission routes and host ranges, what is the risk associated with each pathogen emerging in new species? What are the pathogens that can be prioritised as more-likely to emerge in the future?
Big Data Epidemiology - The Enhanced Infectious Diseases Database (EID2)
Recent years have seen a massive increase in open-access scientific output, both in terms of publications and genomic sequences. For instance, last year alone saw the publication of over 16% of the total number of papers indexed by PubMed, and approximately 20% of the total number of sequences uploaded to Genbank. The sheer volume, not to mention other complexities, of scientific output exceeds the ability of researchers, using traditional methods, to make effective use and assessment of all available findings. The Enhanced Infectious Diseases Database system (EID2) utilises data and text mining tools, with minimal expert input, in order to answer a range of questions such as: 1) What is the host-range of given pathogen/microbe ? 2) What are all the pathogens/microbes of given host? 3) What are all the vector species of certain pathogen? And which hosts do they transmit this pathogen to? 4) What is the geographical range of an organism (host, pathogen or vector)?
In order to provide answers to these questions the EID2 system comprises the following components:
1. Data repositories: EID2 maintains a number of complex data repositories and mapping dictionaries to facilitate interaction discovery and named entity recognition, including: 1) Organisms and their taxonomic lineage relationships (over 1 million organisms to date). 2) Alternative names (e.g. common names, common misspelling, breeds and acronyms), inclusion (AND) and exclusion (NOT) terms for the organisms. 3) Geographical names and hierarchies, including countries, administrative divisions, major cities and natural features. 4) Climate (e.g., temperature and rainfall) and demographic (human and livestock) data for the whole world.
2. Data acquisition layer: EID2 continually retrieves and classifies evidence from two sources: NCBI Nucleotide Sequences database; and PubMed (and soon to include Scopus as a third). Each piece of evidence is then linked to the organisms and geographical location. Sequences are often linked to one “cargo” organism which is either microbe (pathogen) or arthropod vector, one host organism and one location. Publications however are often linked to multiple organisms and locations. One powerful utilisation of EID2 is our ability to quickly extract and filter evidence based on the number of hosts/pathogens/vectors species or locations it mentions. This facilitates other process of EID2, and it enables us to conduct research in other avenues (such as transmission route discovery and co-infection interactions discovery).
3. Interactions discovery pipeline: EID2 extracts three types of interactions from its evidence bases: organism-organism interactions, organism-location interactions and organism-organism-location interactions. (Wardeh et al) provides detailed explanation of the process.
4. EID2 Portal: publically accessible at: https://eid2.liverpool.ac.uk/. The portal enables users to browse through EID2 data, lookup interactions for one or more organisms, and produce tailored maps.
Research groups
Research grants
Predicting animal hosts for future novel coronavirus recombination, and effective strategies for minimising avoidable human-influence driven risk
UK RESEARCH AND INNOVATION
March 2021 - March 2022
Big data approaches to identifying potential sources of emerging pathogens in humans, domesticated animals and crops: NPIF Fellowship for Maya Wardeh
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL, UK RESEARCH AND INNOVATION
November 2017 - September 2021
Big Data approaches to host-pathogen mapping: EID2 - an open-access, taxonomically- and spatially-referenced database of pathogens and their hosts
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
October 2016 - March 2018