Research
The Computational Materials Science and Engineering (CMSE) group applies and develops computational tools to simulate collective phenomena associated with sustainable material fabrication, as well as the properties of solid materials in environments that mimic real systems. The techniques we use are based on molecular and coarse grained particle dynamics simulation methods. We use enhanced sampling techniques and tools from data science to determine the mechanisms and dynamics that describe and control material formation across multiple length and time scales. Additionally, we explore the properties of solids immersed in fluids to understand interfacial processes such as adsorption, charge accumulation and templated assembly. Our research lies at the junction between Chemistry, Physics and Engineering and is applied to explore functional materials, advanced composite materials and formulations for a range of applications.
Our latest publications can be found on Google Scholar.
Please contact us to discuss collaboration opportunities and proposals for fellowship applications. Our current areas of research interest are...
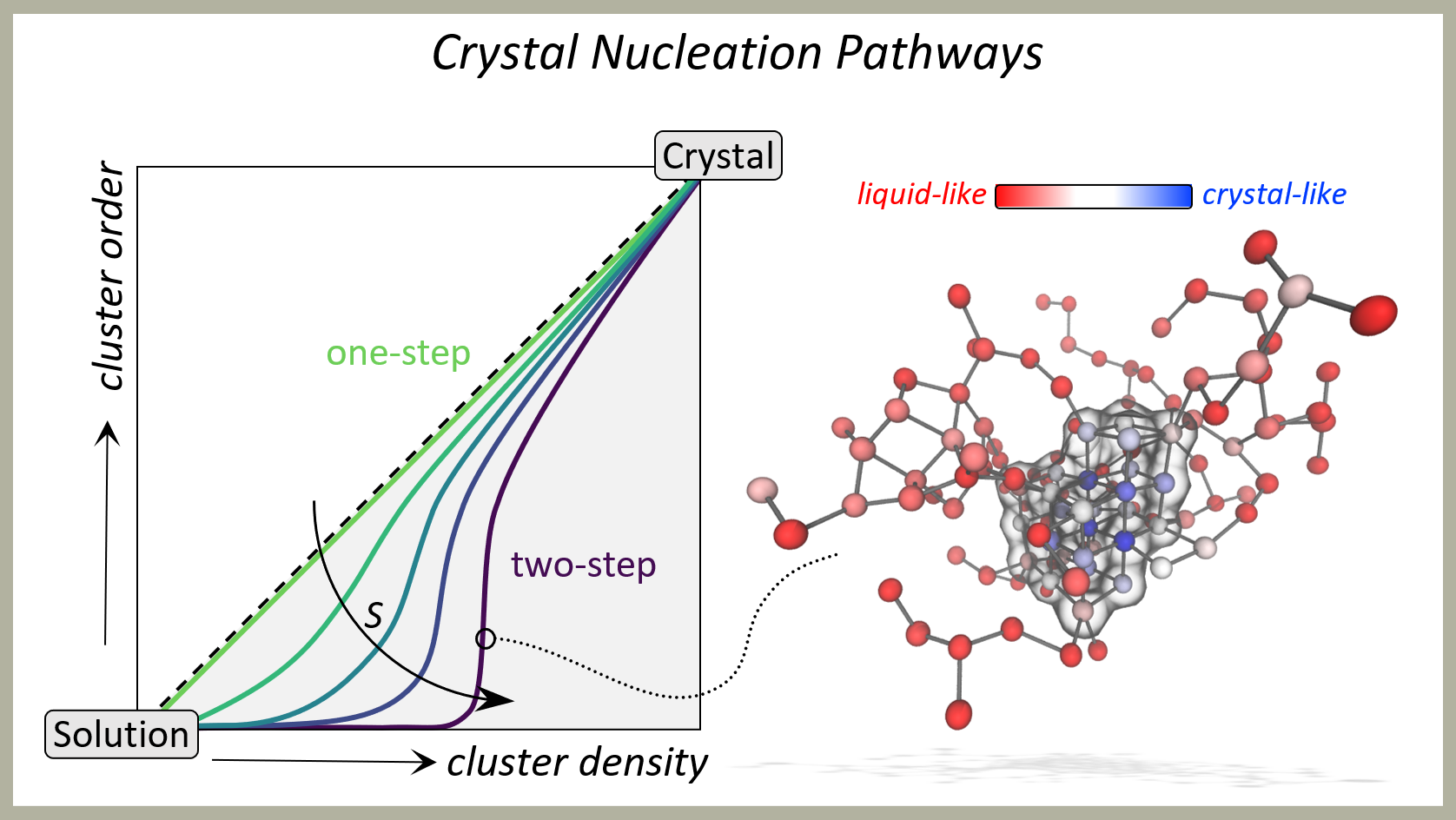
Nucleation and Crystallisation
Thermodynamics govern the behaviour of materials, dictating their transitions between different states depending on prevailing conditions. For instance, liquid droplets can spontaneously form in a high-density gas phase, while salt crystals may emerge from a concentrated solution under favourable conditions. Nucleation marks the initial step in such transformations, the classical theory for which was laid down 150 years ago. However, classical theories often fail to accurately predict nucleation mechanisms and rates observed in practice.
Our research employs molecular and particle dynamics simulations to investigate nucleation characteristics that enable us to evaluate established theories. In the case of crystal nucleation, this demands advanced simulation methods to overcome the inherent challenges of inadequate timescales typically faced in molecular dynamics simulations. Our simulations reveal the microscopic mechanisms and kinetics underlying nucleation and subsequent material growth. By understanding these processes, we aim to design strategies to control nucleation and growth, enabling the targeted formation or inhibition of specific materials.
Our fundamental research holds broad applications across various scientific and engineering fields, offering insights into the development of novel materials and processes.

Self Assembly and Directed Assembly
Self assembly and directed assembly are powerful strategies for creating complex structures and functional materials with tailored properties. In self assembly, molecules or nanoparticles spontaneously organise into ordered structures, which may exhibit structural complexity beyond that of bulk crystals, driven by inherent molecular interactions, such as van der Waals forces.
Directed assembly, on the other hand, involves guiding the assembly process through external cues, such as templating, electric fields and surface patterning. This approach enables precise control over the arrangement and orientation of building blocks, leading to the creation of functional materials with specific properties and functionalities. For example, biology exploits these strategies to construct highly architectured hard and soft tissues.
Our research focuses on exploring the principles of self and directed assembly to engineer functional materials by training models to understand how varying conditions and components influence the formation of diverse material structures and properties. In doing so, we aim to identify design principles to fabricate materials using low-temperature, bottom-up assembly for applications in nanotechnology, photonics, sensors and beyond.
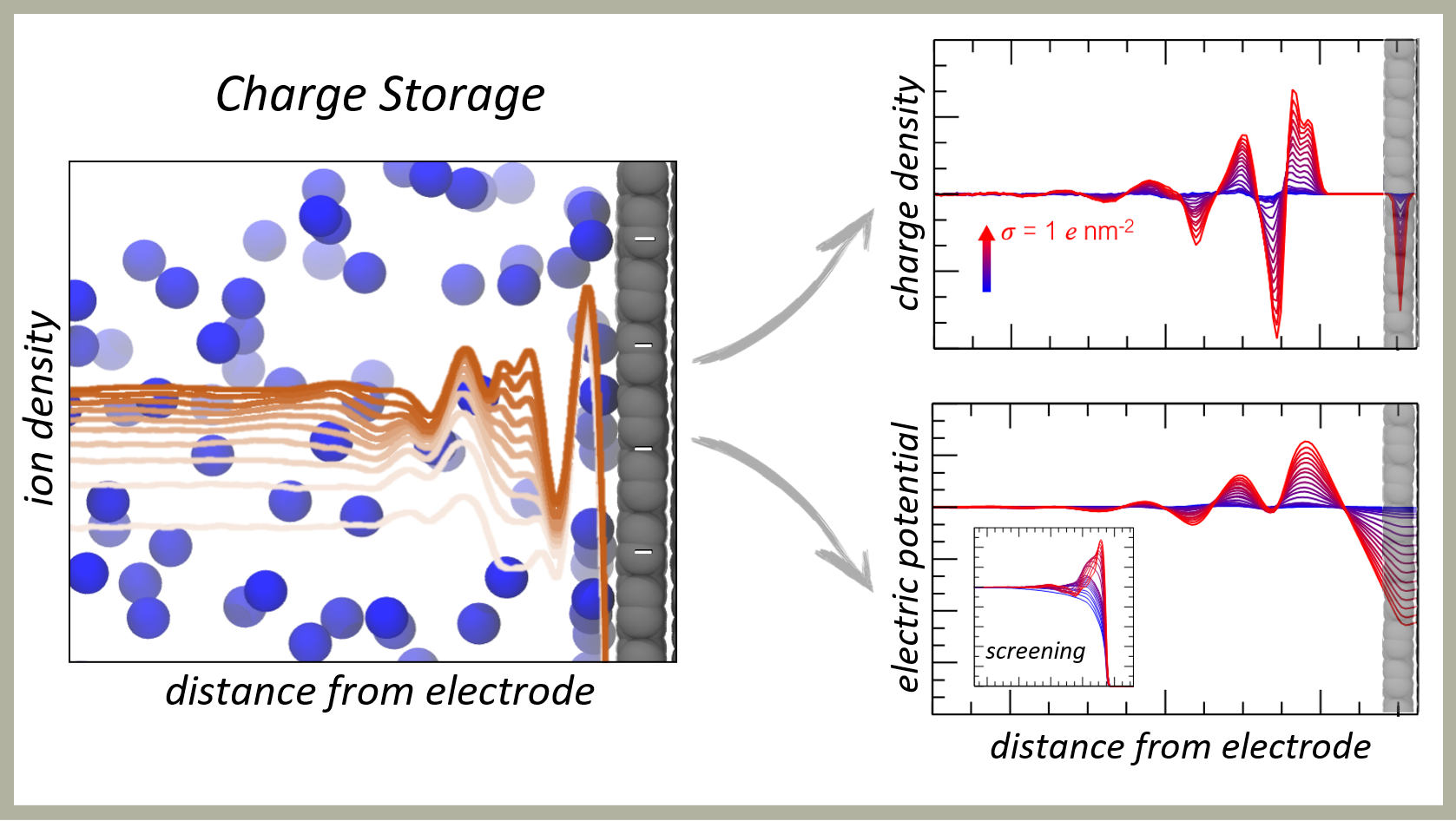
Solid-Liquid Interfaces
Wolfgang Pauli once remarked, 'God made the bulk; surfaces were invented by the devil.' Indeed, characterising surface structures and properties at the atomic scale, particularly when immersed in liquids, poses significant challenges. Solid surfaces exhibit intricate structures that can evolve readily at the nanoscale, influencing the surrounding liquid and giving rise to complex interface regions where important physical and chemical phenomena occur. Characterising these interfaces is crucial for various technological applications in catalysis, separations, sensing, etc.
Our recent focus has been on understanding solid-liquid interfaces in the presence of charged surfaces, particularly relevant for charge storage devices like supercapacitors and batteries. Employing molecular dynamics simulations, we investigate how charged graphite and graphene substrates interact with ions and affect solute and solvent arrangements and mobility at the interface across a wide concentration range. The molecular resolution afforded by the simulations allows us to consider how interfacial structure and electrical properties differ from mean-field theoretical predictions. Additionally, utilising the novel constant chemical potential molecular dynamics method, we quantify changes in solute and solvent activity coefficients near the surface, elucidating interfacial solution thermodynamics.
These findings prompt new inquiries into how surfaces influence particle crystallisation and assembly in liquids, complementing our research in other areas.